
We use electrophysiological, biochemical, and other methods to study the Cystic Fibrosis Transmembrane conductance Regulator.
The McCarty lab is located in the Emory-Children's Center, directly adjacent to the Egleston Children's Hospital of Atlanta and in the heart of a consortium of researchers from many different institutions didicated to understanding and treating CF (the CF@LANTA group). Since CFTR is a chloride channel, the most direct means of measuring its activity is by measuring current across a membrane. Thus, multiple electrophysiological methods are employed in the McCarty lab, including Two-Electrode Voltage Clamp (TEVC), Macropatch, Single-Channel Patch, Ussing Chamber, and Bilayer Recordings. These electrophysiological methods are paired with many biochemical, molecular biology, and computational approaches to obtain a more complete picture of CF.
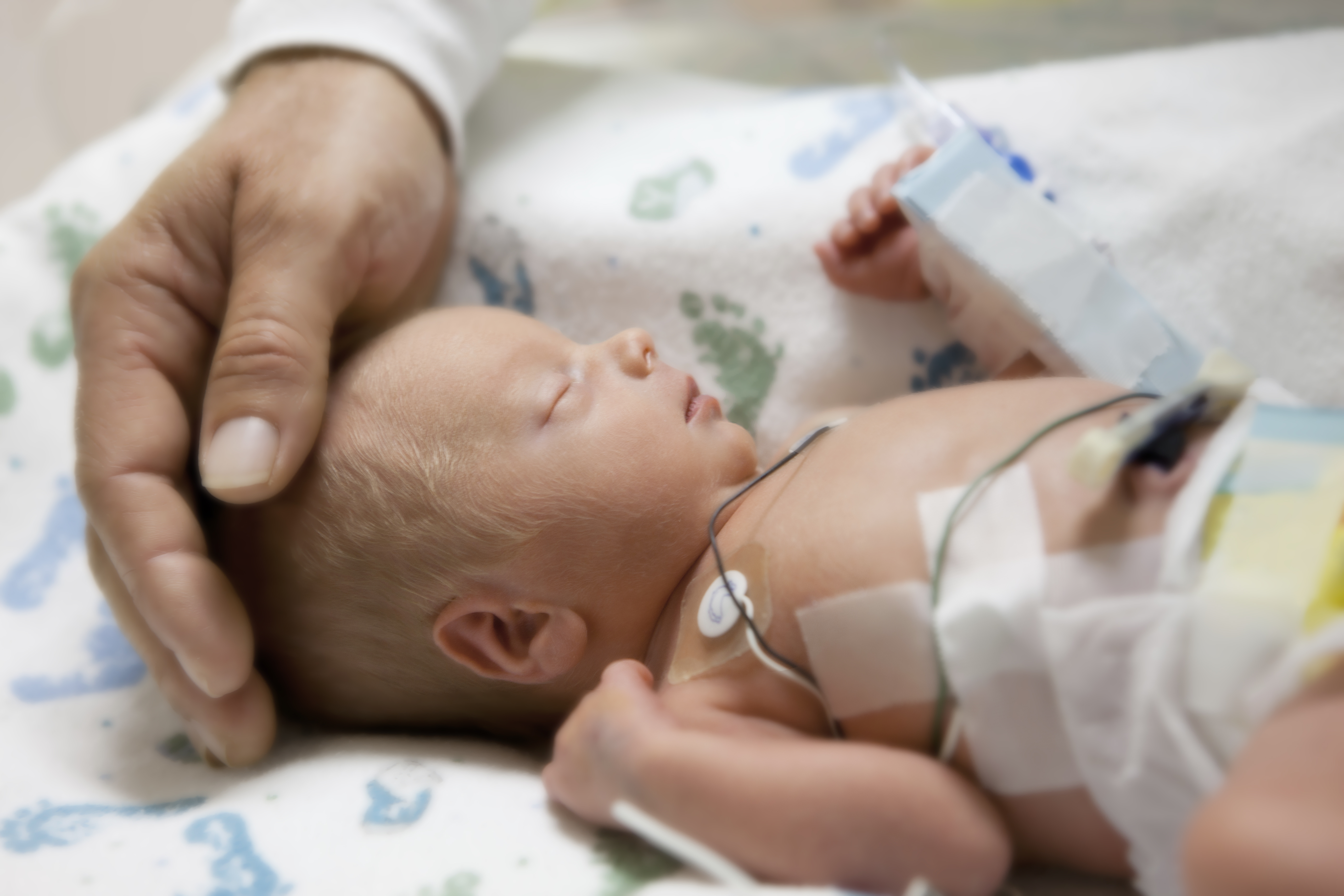
CF-related diabetes (CFRD) impacts nearly half of adult CF patients. CFRD begins to develop early in childhood, and the pathological consequences are severe: CFRD patients have significantly increased frequency of acute pulmonary exacerbations and an accelerated decline in lung function resulting in dramatically shortened lives. Despite the significant impact of CFRD on progression of CF lung disease, the mechanism(s) by which systemic hyperglycemia accelerates acute pulmonary dysfunction in response to infection are unknown. Studies in CF subjects suggest an inability to regulate airway glucose levels. Our extensive preliminary data, including the first demonstration of insulin-dependent glucose uptake by airway cells, point to a direct mechanistic link between expression of mutant CFTR (or loss of CFTR) and dysregulation of glucose homeostasis in airway epithelial cells and mouse lung. Our published data suggest that the combined activities of tight junction proteins and glucose transporters work to create an airway “glucose barrier” that regulates airway glucose levels in normal lungs. In CF, both elements are altered. When barrier dysregulation is coupled with systemic hyperglycemia in CFRD, there is marked elevation of airway glucose levels. We confirmed in our novel mouse model of CFRD that loss of airway glucose control impairs clearance of airway bacteria. Our goals are (1) to identify the mechanisms by which expression of mutant CFTR causes airway glucose dysregulation, (2) to determine the consequences of this derangement on airway bacterial defense, and (3) to determine whether markers of enhanced glycolytic activity in the airways of CF patients may serve as sentinels of converting from normal to impaired glucose tolerance and to CFRD.
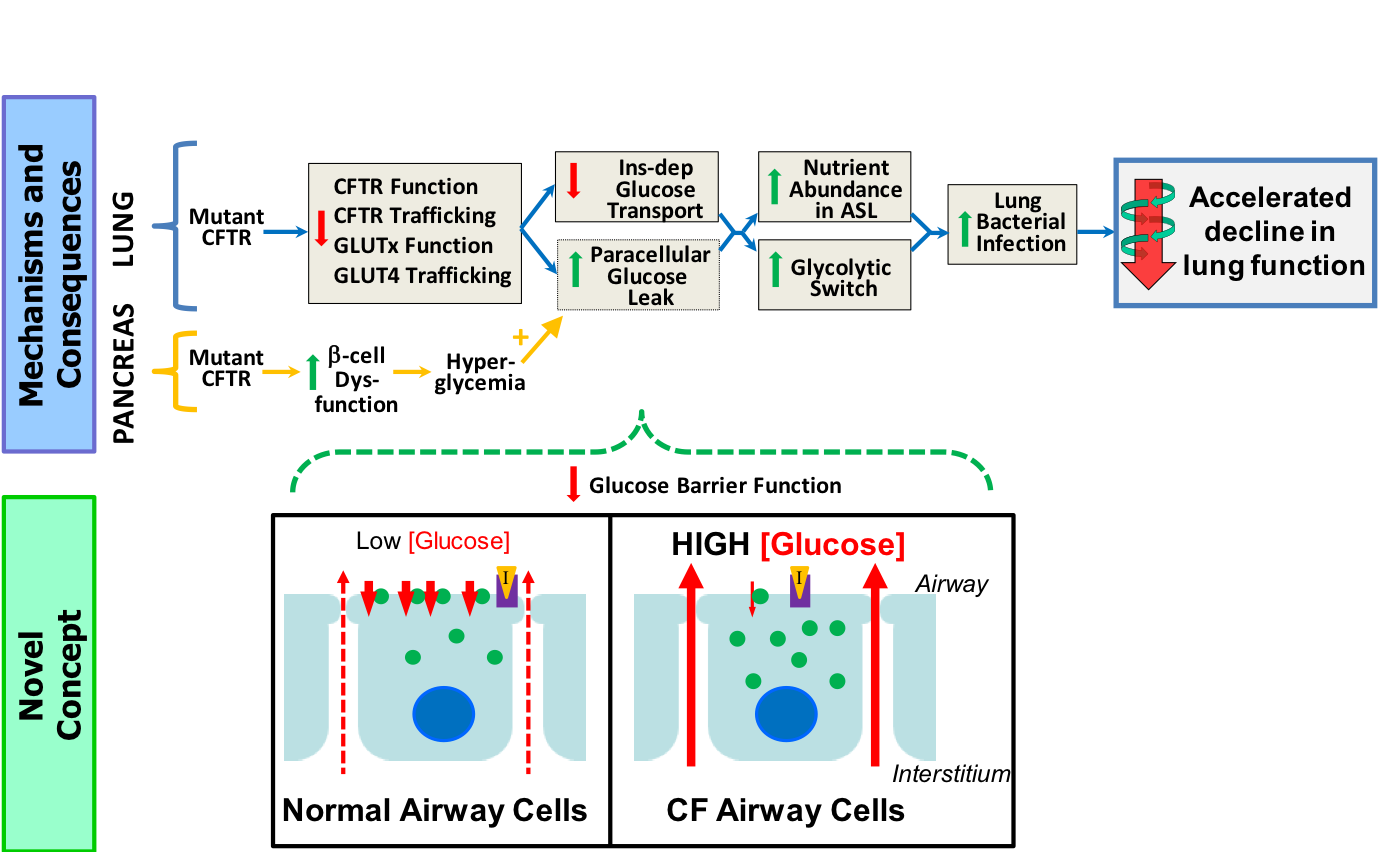
Development of therapeutics as modulators of CFTR took a great leap forward with the identification of the potentiator VX-770 (Ivacaftor/Kalydeco). However, VX-770 does not appear to stimulate all variants of CFTR, and adversely impacts chemical correction of misfolded CFTR proteins; we think we can do better. Other compounds with structures distinctively different from VX-770 can also potentiate CFTR, suggesting that there are features of the protein and its gating mechanism that may be targeted in order to achieve allosteric stimulation of channel activity. However, neither the mechanisms by which these compounds (including Kalydeco) potentiate mutant CFTR nor the site(s) where these compounds bind have been identified. The long-term goal of the proposed research is to determine how small molecules bind to and stimulate CFTR activity in Cystic Fibrosis (CF) patients and patients impacted by some forms of Chronic Obstructive Pulmonary Disease (COPD). This project addresses 3 main challenges regarding potentiation of CFTR:
The mechanism(s): Lack of knowledge about the mechanisms by which CFTR potentiators exert their effect has led to incorrect interpretation of drug efficacy in drug screening campaigns, denying access to potentially life-saving drugs for some patients. We have recently found that the degree of potentiation of CFTR by some compounds exhibits a dependence upon the degree of channel phosphorylation. At the level of mechanism, this observation suggests that at least some of the CFTR potentiators may function as allosteric modulators. At the practical level this observation may require a revision of screening protocols.
The site(s) of action: Unlike some classes of correctors, most CFTR potentiators are thought to act directly on CFTR, based upon mechanistic studies like ours.However, we do not yet know where they bind. A consequence of this knowledge gap is that even now, after the publication of the CFTR structure, we cannot undertake target-based approaches to drug discovery that could identify novel candidates that may bear therapeutic value. This knowledge gap partly reflects the lack of appropriate experimental models to provide clues toward the identification of binding sites.
The drugs: Many different compounds with diverse chemical structures have been found capable of potentiating CFTR. However, these have not been systematically analyzed in a search for common denominators in terms of functional groups. This analysis will allow for the development of pharmacophore models that could be used for ligand-based virtual screening in a search for new, more effective potentiators.
CFTR is a large, polytopic integral membrane protein with multiple functions, most often studied as a chloride ion channel. Loss of CFTR function has been shown by several labs to alter plasma membrane lipids in ways that tie to both increased airway inflammation and decreased innate defenses against bacterial infection. However, the inverse relationship, i.e., the role of lipids in regulating CFTR channel function, has barely been explored. This project focuses on regulation of CFTR by sphingolipids and sphingolipid-mediated signaling. Several CF-relevant bacteria including S. aureus and P. aeruginosa produce virulence factors such as sphingomyelinase (SMase), which breaks down sphingomyelin to ceramide and phosphocholine and has been shown previously to inhibit CFTR by an unknown mechanism. Based on our strong preliminary data (Stauffer et al., 2017), we hypothesize that SMase inhibits CFTR channel activity via a novel mechanism that relies upon lipid-mediated signaling. This may limit the therapeutic potential of CFTR modulators. Our goal is to understand how SMase regulates CFTR activity.

Kerry (left), Kirsten (center), and Nael (right) went to see the Story Collider podcast recording.

Emory CF researchers attend the Great Strides CF Fundraiser Walk.

Beth, Dana, Danny, Brandon, Nael, and some others attend a baseball game at the new Braves Stadium.
The McCarty Lab goes on fun outings!
CURRENT MEMBERS
Staff Scientist: Focuses on electrophysiological methods to understand the structure-functon relationship of CFTR and the mechanism of VX-770 enhancement
Member of McCarty lab since 2001-2005, 2008-Present
Member of McCarty Lab since 2014
Kerry Strickland
Georgia Tech Chemistry graduate student
Member of McCarty Lab since 2014
Kirsten Cottrill
Emory Molecular and Systems Pharmacology graduate student
Member of McCarty Lab since 2017
PREVIOUS MEMBERS
Brandon Stauffer, PhD (graduate student 2010 - 2017)
Danny Infield, PhD (graduate student 2009 - 2016)
Karla Haack, PhD
Stacy Padlov, PhD
Chris Thompson, PhD
Beth Brown, PhD
- Potentiators exert distinct effects on human, murine, and Xenopus CFTR.
- Junctional abnormalities in human airway epithelial cells expressing F508del CFTR.
- Three charged amino acids in extracellular loop 1 are involved in maintaining the outer pore architecture of CFTR.
- Hyperglycemia impedes lung bacterial clearance in a murine model of cystic fibrosis-related diabetes.
- Modeling the conformational changes underlying channel opening in CFTR.
- Two salt bridges differentially contribute to the maintenance of cystic fibrosis transmembrane conductance regulator (CFTR) channel function.
- Differential contribution of TM6 and TM12 to the pore of CFTR identified by three sulfonylurea-based blockers.
- Isolation and characterization of a high affinity peptide inhibitor of ClC-2 chloride channels.
- Evolutionary and functional divergence between the cystic fibrosis transmembrane conductance regulator and related ATP-binding cassette transporters.
- Identification of a region of strong discrimination in the pore of CFTR
A more complete list can be found here.